Mated-pair sequencing e Paired-end Sequencing (Illumina)
DISCLAIMER: O presente texto foi escrito originalmente em Novembro de 2009 e publicado aqui. Reapresento o texto neste blogue a título de consulta e informação. Ao final, adicionei um parágrafo falando sobre a montagem de genomas eucarióticos, trabalho científico que exige computadores e sequenciadores de DNA de última geração. No LAMPADA, trabalhamos com dados de sequenciamento de genomas de humanos (câncer), genomas de diversos pássaros (principalmente beija-flores), genomas de peixes e genomas de plantas. Tudo isso com colaboradores de institutos como o INCA (Carolina Furtado), o Museu Nacional (W Bryan Jennings), o LNCC (Ana Tereza Vasconcelos), o PUC-MG (Daniel Cardoso) e a própria UFRJ (Cássia Sakuragui). Entre em contato conosco caso necessite de maiores informações sobre o sequenciamento de genomas humanos, animais ou vegetais.
===
Em palestras e também no sítio da internet da empresa Illumina sempre nos deparamos com duas "formas" diferentes de se realizar o sequenciamento NGS: (i) o paired-end sequencing e o (ii) o mated-pair sequencing. Depois de muito discutir com diversos colegas sobre as diferenças ou semelhanças entre os métodos, finalmente encontramos explicações mais específicas sobre o que significa sequenciar nossa amostra através de um ou outro método -- de acordo com os conceitos definidos pela Illumina.
.: História da genômica :.
Vale lembrar aqui a origem dos conceitos de mated-pair e paired-end sequencing, isso nos levará também à história da genômica e do projeto genoma humano.
Em 1992, J Craig Venter publica cerca de 300 sequências parciais de genes (ESTs) humanos expressos no cérebro [1]. À época o GenBank possuia cerca de 3000 genes humanos e numa tacada só Venter publicava uma quantidade de informação que era comparável a 10% do que todos os cientistas do mundo haviam feito até a época. Catapultado ao estrelato da ciência, por volta de 1998 o cientista e empreendedor americano diz ao mundo que estaria pronto para sequenciar o genoma humano inteiro. Tendo fundado a empresa Celera Genomics, Venter promete sequenciar rapidamente o genoma de nossa espécie e, é claro, tem o interesse em vender seu trabalho para empresas farmacêuticas, interessadas no potencial biotecnológico do conhecimento do genoma. Todas elas queriam ter acesso, o quanto antes, às informações sobre os genes e proteínas humanos que poderiam permitir a produção de novas drogas para as mais diversas doenças. Venter promete terminar o genoma humano em prazo recorde e sugere inclusive aos pesquisadores do agora chamado "consórcio público" que poderiam parar de sequenciar os dados humanos, posto que ele seria capaz de fazê-lo com qualidade e eficiência, liberando-o publicamente para a pesquisa científica depois de algum tempo -- sabe-se lá quanto -- em que as empresas farmacêuticas teriam exclusividade sobre os dados. Mas como deixar o maior empreendimento humano do século nas mãos de cientistas com ideal capitalista? O consórcio público gritou: Não!, e então começou a corrida pelo genoma. Quem o produziria primeiro e com melhor qualidade? O consórcio público ou o privado?

O polêmico e brilhante pesquisador americano J Craig Venter, nascido em 1946. Sua história pessoal confunde-se com a história da ciência genômica.
Muitos duvidavam que Venter fosse capaz de sequenciar um genoma assim tão grande de eucariotos, principalmente devido ao fato de que sua estratégia não levava em consideração os mapas de ligação e era baseada principalmente na técnica chamada de whole-genome shotgun -- onde o genoma é picotado em pedacinhos e depois montado. Sequenciar o genoma humano era um problema muito mais complexo do que sequenciar o genoma de uma bactéria, que Venter já havia provado saber fazer (em 1995) com o genoma da Haemophilus influenzae, o primeiro genoma de um organismo de vida livre já publicado [2]. Os pesquisadores então duvidavam tanto de sua capacidade e equipe técnica quanto da viabilidade de sequenciar o genoma numa época onde quase nenhum genoma ainda havia sido "conquistado". Para provar entretanto que era capaz de fazê-lo, Venter foi buscar o atestado de eficácia do seu método ao sequenciar o genoma do organismo modelo mais clássico da genética: a mosca-das-frutas (Drosophila melanogaster) [3].

A tabela 2 do paper que descreve o genoma da mosca-das-frutas [2]. A tabela mostra o tamanho das 3 bibliotecas produzidar e o número de clones sequenciados. O sequenciamento de extremidades de clones contendo grandes insertos facilita a posterior montagem dos segmentos através de técnicas de bioinformática.
O segredo que Venter encontrou para sequenciar genomas grandes era, ao invés de realizar o chamado whole-genome shotgun -- onde o genoma era todo partido em pedaços do mesmo tamanho e sequenciados a partir de primers localizados no vetor de clonagem -- usar uma estratégia similar. Então, ao invés de clonar todos os fragmentos do mesmo tamanho, Venter clonou fragmentos de tamanhos diferentes e seu braço direito Mark Adams, criou programas de bioinformática capazes de levar em consideração diferentes tamanhos de fragmentos entre duas sequências de forma a montar o genoma com eficácia.
Com a publicação do genoma de Drosophila, Venter mostrou que tinha know-how para produzir genomas ainda maiores e mais complexos, como o genoma humano. De fato, o rascunho do genoma humano foi publicado em 2001 numa publicação conjunta dos dados públicos (na Nature) e dos dados da Celera (publicados na Science).
.: Mated-pairs X Paired-ends :.
A idéia de produzir então bibliotecas genômicas de diferentes tamanhos e sequenciar extremidades de insertos inseridos nestas bibliotecas para depois montá-los -- esperando que insertos de diferentes clones se sobreponham -- parece ter vindo da aventura de Venter et al. (2000) ao sequenciar o genoma de Drosophila, mostrando ao mundo que o caminho estava aberto para a sequência completa do genoma humano. De fato, a estratégia era bastante lógica e elegante; e a montagem do genoma parece ter sido mais fácil do que se supunha anteriormente.
O conceito dos mated-pairs ou pair-ends, portanto, tem a ver com o fato de que gera-se fragmentos (insertos) de bibliotecas genômicas com diferentes tamanhos e assim sequencia-se-os de um lado e do outro. Na figura mostrada, vemos que para montar o genoma de drosófila, Venter utilizou três bibliotecas e sequenciou clones da extremidade de insertos contendo 2Kb, 10Kb e 130Kb. A idéia embutida neste processo está relacionada ao fato de que existem repetições nos genomas de eucariotos e às vezes convém ter uma sequência cuja distância de uma primeira é conhecida; assim, no caso desta primeira mapear em uma região de repetição, seria agora possível ancorar a sequência do outro lado do clone em algum lugar conhecido e de cópia única no genoma. Como também o sequenciamento dos reads acontece de forma aleatória com relação à posição deles no genoma, a utilização de bibliotecas de diferentes tamanho pode funcionar como um controle positivo do estudo, garantindo que sequências montadas em 2Kb estão a 10Kb quando observadas aquelas oriundas da segunda biblioteca. De fato, esta é uma técnica elegante e eficiente para facilitar a montagem do genoma. É claro, entretanto, que ela necessita de algoritmos mais elaborados de montagem genômica. Adams certamente não deixa por menos.
Hoje em dia existem diversos programas que montam genomas baseando em dados de pares de sequências cujo tamanho entre eles seja conhecido. O programa define um tamanho máximo ou mínimo de sequência que deve estar ali entre aqueles 2Kb, digamos, e monta as sequências com relação a outras. O buraco (gap) que existe entre duas sequências é preenchido por dados do sequenciamento de outras reads.
No caso do Illumina GAII, por exemplo, podemos ter uma cobertura de cerca de 100x do genoma de uma bactéria para cada canal da flowcell (são 8 canais por placa). Com estes cerca de 600Mb produzidos num único canal normalmente somos capazes de amostrar 100 vezes cada pedaço do genoma de uma bactéria (considerando um genoma bacteriano de 6Mb). Esta alta cobertura praticamente garante que o genoma será fechado e terá alta qualidade em todas suas posições nucleotídicas. De fato, uma cobertura de 8-10x normalmente é suficiente para produzir um genoma completo de qualquer espécie. Isso significa que, se o genoma tem X pares de base, é preciso sequenciar 8 a 10 vezes (8X) o tamanho do genoma para garantir que cada parte será amostrada ao menos uma única vez -- já que a região sequenciada consiste em uma parte aleatória do genoma do organismo de interesse. Os números 8-a-10 vêm de análises estatísticas realizadas sobre o assunto (Lander e Waterman, 1988) [5][6].
Quando Venter e seus colaboradores sequenciaram o inserto pelos dois lados, essa técnica ficou conhecida como mated-pair ou paired-end sequencing. Tanto que quando fui apresentado a esses termos num contexto NGS-Illumina, tive a impressão de que os conceitos eram sinônimos. Entretanto, com relação a este sequenciador e esta empresa, foram criados realmente dois conceitos novos que representam coisas diferentes.
Basicamente a diferença é a seguinte:
- paired-end: significa o sequenciamento a partir das duas extremidades para fragmentos de DNA com o tamanho entre 200-500bp;
- mated-pairs: significa o sequenciamento a partir de duas extremidades de fragmentos de DNA com o tamanho entre 2-5Kb. (De fato, o sequenciamento não se dá à partir do fragmento per se, mas a partir da reunião de suas extremidades e sequenciamento de uma molécula contendo apenas essas extremidades da molécula.)
A questão da diferenciação está relacionada ao fato de que enquanto no caso dos paired-ends é necessário ligar adaptadores dos dois lados da sequência e ligá-la à flowcell (placa de vidro), no caso dos mated-pairs (quando o tamanho do inserto é grande) torna-se necessário realizar um protocolo especial antes da ligação, de forma a unir as pontas e permitir o sequenciamento das extremidades numa sequência pequena.
.: Paired-end sequencing :.
O sequenciador Illumina permite que os pesquisadores obtenham sequências de ambas extremidades de um fragmento de DNA. Para isso é preciso simplesmente ligar adaptadores dos dois lados da sequência e sequenciá-los da forma padrão como é feita pelo Illumina. Uma corrida de paired-end é capaz de produzir 2x75bp em até 200 milhões de reads.

A técnica de sequenciamento das extremidades de um inserto contendo entre 200-500bp é chamada de paired-end sequencing pela empresa Illumina
.: Mated-pair sequencing :.
Caso desejemos, entretanto, sequenciar extremidades de fragmentos maiores, é preciso que realizemos alguns truques do arsenal da biologia molecular. O kit da Illumina, entretanto, permite a realização da técnica de maneira simples.
Como parece não ser possível ligar à flowcell (placa de vidro) sequências muito grandes de DNA, os gurus da Illumina inventaram uma técnica eficiente para selecionar extremidades de clones grandes e ligá-los de forma a permitir o sequenciamento de suas extremidades.
O truque de biologia molecular realizado pelos técnicos da Illumina consiste nos seguintes passos:
- Corta-se o DNA e purifica-se moléculas do tamanho desejado
- Liga-se biotina na extremidade das moléculas
- Circulariza-se o DNA
- Realiza-se a purificação do DNA circularizado
- Corta-se o DNA circular em fragmentos de 400-600bp (esse tamanho de fragmento já pode ser ligado à flowcell)
- Seleciona-se entre os fragmentos cortados, aqueles que possuem biotina
- Liga-se os adaptadores nas sequências com biotina
- Sequencia-se a partir dos dois lados (paired-end sequencing)
O que se faz, portanto, é reunir as extremidades que se deseja sequenciar através da circularização do genoma e então retirar as partes que não correspondam às extremidades. Apenas as regiões da extremidade serão sequenciadas. É uma técnica interessante e elegante, embora exija mais trabalho técnico na bancada.

A técnica de sequenciamento das extremidades de um inserto contendo entre 2-5Kb é chamada de mated-pair sequencing pela empresa Illumina
.: E como sequenciamos extremidades de clones entre 500bp-2Kb ou maiores do que 5Kb? :.
No sítio da Illumina não está claro o que fazer caso se deseje sequenciar clones que tenham um tamanho intermediário entre 500bp e 2Kb. É teoricamente possível que o sequenciamento mais simples por paired-ends funcione, embora talvez não dê resultados muito satisfatórios devido a problemas na bridge-PCR. É teoricamente possível supor também que insertos maiores do que 5Kb poderiam funcionar através da técnica de mated-pairs para produzir as sequências contendo apenas as extremidades das sequências. Entretanto talvez seja preciso entrar em contato direto com a empresa para verificar mais informações a respeito destes detalhes.
.: Montando genomas eucariotos completos :. (adicionado em 16/09/2014)
Para realizar a montagem de genomas eucariotos completos, necessita-se, necessariamente do sequenciamento do genoma usando diferentes bibliotecas, tanto bibliotecas paired-ends quanto bibliotecas mated-pairs. Normalmente sequencia-se pelo menos uma ou duas lanes inteiras de illumina usando bibliotecas paired-end e pelo menos outra lane contendo uma biblioteca mated-pair. Com isso é possível conseguir um rascunho do genoma a ser montado com programas como o SOAP-denovo, AllPaths, Abyss, Velvet, dentre outros.
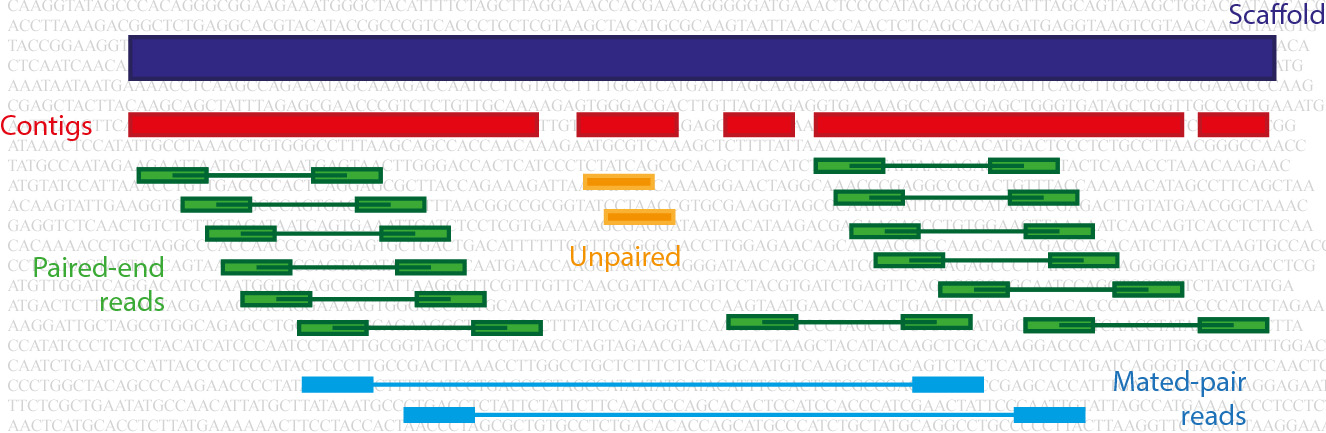
Tais programas permitem que o usuário entre com as informações das distâncias entre os inserto sequenciados. Os programas montam os genomas em duas etapas. Primeiro realizam o processo de contigação (contigging), onde separam as sequências por kmers e montam grafos de kmers completos e contínuos, onde as sequências são concatenadas através do compartilhamento de kmers (palavras de tamanho N, normalmente N=23). Na segunda etapa de esqueletação (scaffolding), os programas levam em consideração a distância entre os insertos do mated-pair e do pair-end e adicionam tantas letras "N" quantas forem necessárias para preencher o tamanho esperado, reunindo então contigs entre si para formar os esqueletos de sequência (ou scaffolds). Tais esqueletos contêm contigs dos quais tenha sido possível uni-los através da informação do mate pair.
Uma vez montados os genomas, parte-se para etapa da predição gênica, onde os genes devem ser localizados e identificados. Em uma primeira versão do genoma, espera-se que os genes sejam descobertos ainda de forma preliminar e parcial. Apenas com novas versões do genoma, novos sequenciamentos usando outras tecnologias (Pacific Biosciences, 454, Ion Torrent) é que se torna possível melhorar a montagem e anotação do genoma.
De qualquer forma, uma publicação do genoma parcial ou sua primeira versão é de inestimável ajuda para grupos que estudem o animal. Principalmente, elas já têm regiões disponíveis para fazer PCR e identificar o animal, além de marcadores possíveis para realizar paternidade e diversos outros tipos de estudo que se tornam possíveis quando o genoma de um determinado micróbio, animal ou vegetal está disponível para o estudo.
A figura a seguir mostra um exemplo de montagem de Contigs e Scaffolds usando leituras paired-end e mate-paired. Créditos da figura: Francisco Prosdocimi, 2014.
====
[1] Adams MD, Dubnick M, Kerlavage AR, Moreno R, Kelley JM, Utterback TR, Nagle JW, Fields C, Venter JC. Sequence identification of 2,375 human brain genes. Nature. 1992 Feb 13;355(6361):632-4.
[2] Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995 Jul 28;269(5223):496-512.
[3] Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Sidén-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, WoodageT, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. The genome sequence of Drosophila melanogaster. Science. 2000 Mar 24;287(5461):2185-95.
[4] http://www.hhmi.org/news/rubin3.html
[5] Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988 Apr;2(3):231-9.
[6] Genome sequence assembly primer. Center for Bioinformatics and Computational Biology. University of Maryland.
[7] Paired-end sequencing by Illumina: http://www.illumina.com/technology/paired_end_sequencing_assay.ilmn
[8] Mated-pair sequencing by Illumina: http://www.illumina.com/technology/mate_pair_sequencing_assay.ilmn
 Biólogo, Mestre em Genética, Doutor em Bioinformática. Trabalha com montagem e anotação de genomas animais, genômica e transcriptômica comparativa, filogenômica e genética de ...
Biólogo, Mestre em Genética, Doutor em Bioinformática. Trabalha com montagem e anotação de genomas animais, genômica e transcriptômica comparativa, filogenômica e genética de ...